UNSW Embryology |
|
|||||
Major Abnormalities by Systems |
|
|||||
|
|
|
|||||
|
Page
Links |
Page
2 | Abnormalities
| OMIM
| Self
Assessment Questions |
Medline |
|||||
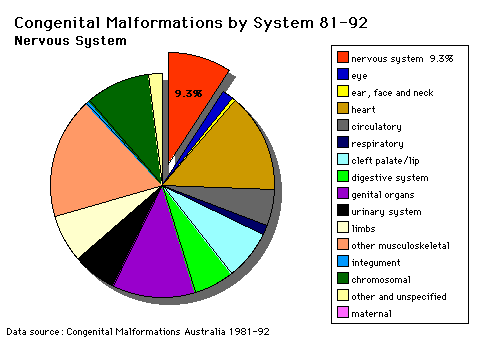
Nervous System |
||||||
|
|
||||||
SPINA BIFIDA (MENINGOMYELOCELE)The main objective in using Spina Bifida as a teaching model is to stress the main features in an integrated approach to the total care of the Spina Bifida patient. Questions related to Spina Bifida (a) Describe the possible processes involved in the abnormal development of the C.N.S. in Spina Bifida. At what time in development would these abnormal events occur? (b) What is amniocentesis? Would this procedure be useful in the diagnosis of Spina Bifida? Give reasons for your answer. (c) What is the effect of the lesion on the bladder, bowel and limbs? Describe the differences on these areas between lesions at the following levels: a. Cervical (d) If the lesion is at higher thoracic levels, why would the prognosis for the patient be poor? (e) Compare and contrast the following: (i) Transection |
||||||
Brown-Sequard Syndrome (Hemisection) |
||||||
Spina Bifida |
||||||
Senses |
|
|||||
|
|
||||||
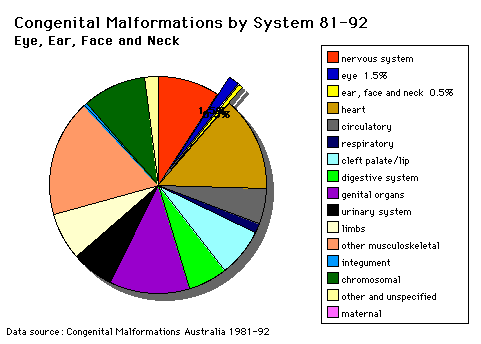
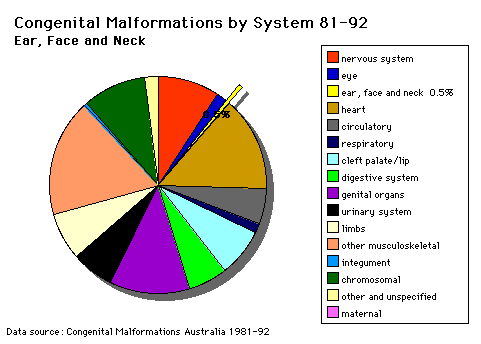
Head and Neck |
|
|||||
|
|
||||||
|
The malformations CLEFT LIP and palate and PIERRE ROBIN SYNDROME during the lecture series and demonstration materials will be provided during the practical class. Cleft lip and palate develop between the 4th and 8th week of gestation and is dominated by changes resulting in the formation of the nose. Palatal development occurs between the 7th and 12th week of gestation and is divided into the formation of the primary palate (prolabium), premaxilla and cartilaginous septum) and formation of the secondary palate (hard and soft palate). In the treatment and repair of cleft lip the following results are hopefully achieved: (i) a symmetrical lip The major objectives of the repair of the cleft palate includes construction of a competent, functioning and watertight valve at the junction of the soft palate and pharynx; repair is performed early enough to allow the child to begin speech with a functioning velopharyngeal valve. Presentation of normal hearing must also be maintained along with normal development and bone growth in the central facial region along with a functional and attractive dentition. |
||||||
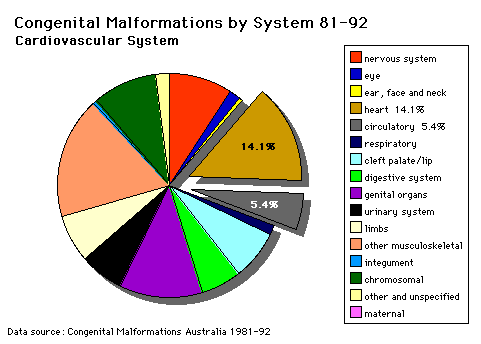
Cardiovascular System |
|
|||||
|
|
||||||
|
PATENT DUCTUS ARTERIOSUS (P.D.A.) OMIM Database Entry- Patent Ductus Arteriosus (about this entry) |
||||||
|
ATRIAL SEPTAL DEFECT (A.S.D.) OMIM Database Entry- Atrial Septal Defect (about this entry) |
||||||
|
TETRALOGY OF FALLOT (i) ventricular septal defect |
||||||
Ventricular septal defect
Congenital Malformations Australia 1981-1992 P. Lancaster and E. Pedisich ISSN 1321-8352 |
||||||
Transposition of Great VesselsCharacterized by aorta arising from right ventricle and pulmonary artery from the left ventricle
Congenital Malformations Australia 1981-1992 P. Lancaster and E. Pedisich ISSN 1321-8352 |
||||||
Hypoplastic Left HeartCharacterized by obstructive valvular and vascular lesion of the left side of the heart.
Congenital Malformations Australia 1981-1992 P. Lancaster and E. Pedisich ISSN 1321-8352 |
||||||
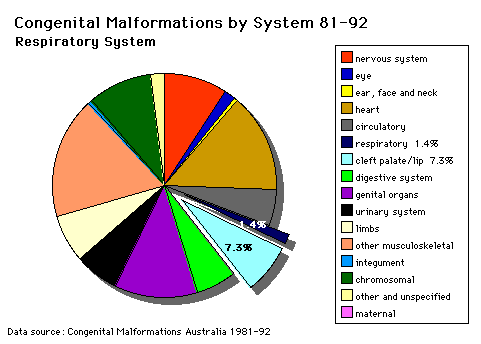
Respiratory System |
|
|||||
|
|
||||||
TRACHEO-OESOPHAGEAL FISTULA (OESOPHAGEAL ATRESIA)OMIM Database Entry- Tracheosophageal Fistula OESOPHAGEAL ATRESIA- with or without tracheo-oesophageal fistula
Summary Oesophageal atresia as suggested by (a) Polyhydramnios Management 1. Operative closure of the trachea end of the fistula. Associated Malformations (a) Stratified squamous epithelium and columnar epithelia (mixtures of tracheal and oesophageal epithelial) are found in the trachea and oesophagus. Questions (a) The failure of what embryological process might give rise to this malformation?
|
||||||
LOBAR EMPHYSEMA (Overinflated lung)OMIM Database Entry- Lobar Emphysema Description of lesion - there is a congenital deficiency of cartilage in the left upper lobe bronchus. The anatomical nature of the lesion was demonstrated by autopsy specimens and usually reveals no abnormality other than cartilage deficiency throughout the lobe. Summary (a) Respiration is normal at first Management The affected lobe must be resected. |
||||||
RESPIRATORY DISTRESS SYNDROME IN THE NEWBORN (Hyaline Membrane Disease)Management 1. R.D.S. is normally treated conservatively with 02, intravenous bicarbonate and general supportive measures but the mortality rate is high in more premature infants. At autopsy the principle change to be seen is an eosinophilic hyaline membrane filling the alveoli. |
||||||
CONGENITAL DIAPHRAGMATIC HERNIAFailure of the pleuroperitoneal foramen (foramen of Bochdalek) to close allows viscera into thorax. Intestine, stomach or spleen can enter the pleural cavity, compressing the lung.
|
||||||
AZYGOS LOBECommon condition (0.5% of population). The right lung upper lobe expands either side of the posterior cardinal. Management Condition is generally harmless. |
||||||
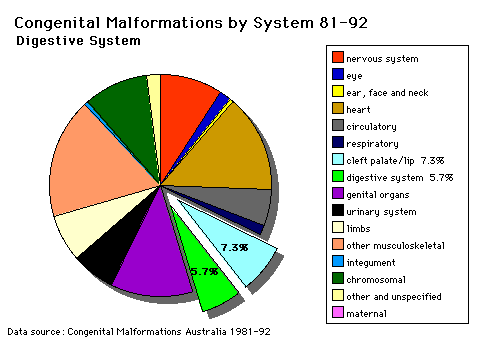
Digestive System |
|
|||||
|
|
||||||
|
I. INTESTINAL MALROTATION What abnormal embryological processes could interfere with normal rotation and fixation of the gut? OMIM Database Entry- Volvulus of Midgut (about this entry) II. SITUS INVERSUS VISCERA Disturbance of the lateralisation of the liver may produce transposition of some or all of the foregut and its derivatives. 1. Stomach Also in situs inversus the anatomical relations of the duodenum, pancreas, bile ducts and portal veins may be reversed or disordered. OMIM Database Entry- Situs Invertus (about this entry) III. MECKEL'S DIVERTICULUM OMIM Database Entry- Meckel's Diverticulum (about this entry) IV. HIRSCHSPRUNG'S DISEASE (INTESTINAL AGANGLIONOSIS) OMIM Database Entry- Hirschsprung's Disease (about this entry) OMIM Database Entry- Hirschsprung's Disease 2 (about this entry) V. GASTROINTESTINAL ABNORMALITIES, MULTIPLE OMIM Database Entry- Multiple GIT Abnormalities (about this entry) |
||||||
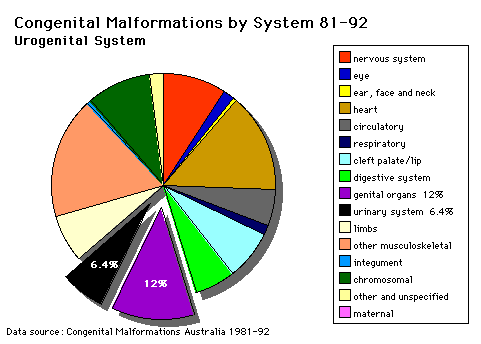
Urogenital System |
|
|||||
|
|
||||||
|
Note that upper G.I.T. obstruction is associated with POLYHYDRAMNIOS whereas failure of fetal micturition is associated with OLIGOHYDRAMNIOS with consequent firm uterine moulding on the fetus, leading to facial, locomotor and palatal deformities.The "figures" referred to below are on posters in the practical classroom. |
||||||
RENAL AGENESIS(a) In the complete form the child is not viable and the child dies within a few days of birth. (b) Features associated with this anomaly are:- (i)Oligohydramnios (ii) Amnion nodosum (small warty amnion with accretions of squamous cells on the inner wall). This is tangible evidence for oligohydramnios. (iii) Facial deformities: This results from uterine moulding around the head. (Figure 1). The ears are low slung and simple, the mandible is small, the nose flattened and the eyes exhibit Pre-epicanthic folds (Figure 2). This is a horseshoe shaped flap of skin from the upper lid to the cheek in front of the epicanthus. (Downs syndrome has an epicanthic fold). Note that the genesis is occasionally incomplete allowing survival (e.g.) Figure 2. Causal factors are largely unknown although there is some familial predisposition. |
||||||
POLYCYSTIC KIDNEYSThis is a diffuse cystic malformation of both kidneys with cystic malformations of the liver and lung etc. often being associated. There is often familial disposition with this malformation. There are TWO types (i) Infantile (inconsistent with prolonged survival) (ii) Adult (less severe and allows survival) |
||||||
MULTICYSTIC KIDNEY(i) This is non familial and is produced by atresia of a ureter (ii) It is always unilateral (iii) There is no functional kidney tissue present in the kidney (iv) The kidney is replaced by a multiocular cyst. |
||||||
URINARY TRACT OUTFLOW OBSTRUCTION1. (e.g.) Urethral valves (Figure 9) - This figure is a micturating urethrogram and shows a valve obstruction (arrow) with dilatation of the urethra between the valve and the bladder. This type of pre-natal obstruction produces gross hydronephrosis and hydroureter before birth. Figure 10 shows gross dilatation of the pelves and ureters. There is extensive destruction of renal tissue. 2. Congenital Hydronephrosis is usually due to partial obstruction at the pelvi-ureteric junction (Figure 11). The pelvis is shown to be grossly dilated and there is extensive renal damage before birth. *This may be familial, may be lateral, and is most commonly an intrinsic defect in the wall of the ureter (structural or functional). The less severe cases may be salvaged by reconstruction of the pelvi-ureteric junction. |
||||||
PRUNE BELLY SYNDROME (Triad Syndrome)The Triad is (i) Agenesis of abdominal wall muscles (ii) Bladder outflow obstruction (iii) Bilateral undescended testes *The causes of this malformation are little known, but maternal therapy with Oestrogens in the first trimester has been implicated frequently. Question: Does oestrogen possibly inhibit the development of the male bladder outflow and genital system? In some cases there are vestiges of muscle in the abdominal wall and it is not known whether this represents (a) destruction of muscle, or (b) failure of development of muscle Figures 12 and 13 show a typical prune belly. Survival of the prune belly child depends on the number of functioning remaining nephrons at birth and the operability of the obstruction. |
||||||
HORSESHOE KIDNEYIn the horseshoe kidney there is fusion of the lower poles of the kidney.During migration from the sacral region the two metanephric blastemas can come into contact as shown in Figure 14 mainly at the lower pole. The ureters pass in front of the zone of fusion of the kidneys. The kidneys and ureters usually function adequately but there is an increased incidence of upper urinary tract obstruction or infection. |
||||||
EXSTROPHY OF THE BLADDERManagement of Bladder Exstrophy The deformity is non-familial, of no known cause and is obvious at birth. This malformation produced incontinence. The surgical reconstruction is complex and requires simultaneous repair of the bony pelvis and covering of the bladder and bladder neck. The epispadiac urethra is reconstructed later. (Ref. Snell, Clinical Embryology, p. 215, fig. 15-16). |
||||||
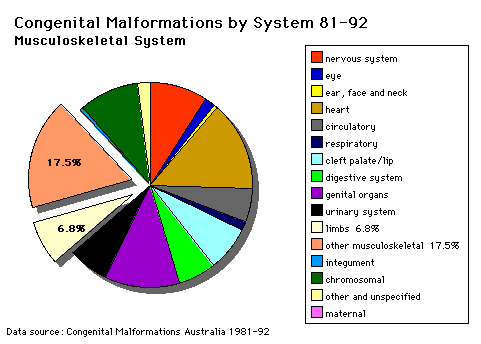
Musculoskeletal System |
|
|||||
|
|
||||||
|
Clinical Introduction: 1. Normal growth and development of the limb requires (a) normal cell numbers; (b) normal locomotor elements, e.g. bone, joint, muscle; (c) normal blood and nerve supply. 2. Retardation of limb growth is produced by (a) deficient nerve supply; (b) impaired blood supply or (c) systemic abnormalities affecting the growth plate. 3. Increased limb growth is produced by some nerve malformations and by increased blood supply to the growth plate. The "figures" referred to below are on posters in the practical classroom. |
||||||
|
CONGENITAL DISLOCATION OF THE HIP (C.D.H.) OMIM Database Entry- Congenital Hip Dislocation Introduction (Instability: 1:60 at birth; 1:240 at 1 wk: Dislocation untreated; 1:700). (a) There is originally a congenital instability of the hip which later dislocates by muscle pulls or gravity if untreated. (b) There is familial predisposition for this problem and female predominance. (c) Growth of the femoral head, acetabulum and innominate bone are delayed until the femoral head fits firmly into the acetabulum. Mechanisms of Production (a) There is familial displasia of the hip. Treatment Treatment must be instituted early to avoid a growth deformity of the hip. To ensure there is no instability, infants are tested at birth for hip stability and unstable hip children are nursed in the Frog Position (abducted hip posture). Photo CDH1 Pawich Brace Photo CDH2 Frog Plaster Delay in treatment leads to frank dislocation of the hip (the femoral head comes out of joint), and there is a shallow acetabulum and a small femoral head. See Photo CDH3. If this condition is allowed to occur an operation may be necessary to produce a more horizontal roof to the acetabulum and produce hip stability. See Photo CDH4. Posterior dislocation of the hip produces flexion deformities of the hip with compensatory Lordosis - exaggerated lumbar curvature. See Photo CDH5 (both female). Questions 1. In CDH5 the smaller child on the right shows Trendelenberg's Sign - as she raises her right foot the right side of the pelvis lowers instead of raising. In the normal patient the hip rises when the ipsilateral foot is raised from the ground. What muscle is chiefly responsible for the normal tilting of the hip? 2. What conditions may give rise to Trendelenberg's Sign? |
||||||
|
ARTHROGRYPOSIS (Multiplex Congenita) Rare Severe cases are characterised by multiple deformities at birth with gross stiffening of joints and hypotonia or wasting of muscles. Photo AG1. Such a stiff fetus frequently sustains fractures before or during delivery. AG1 has had a fractured right humerus. Photo AG2 shows deformities originally thought to be joints, then joints and muscles then finally innervation was also implicated. Photo AG3 shows normal and abnormal muscles in close proximity. Variations in the degree of severity of joint deformity are expressions of varying degrees of muscular and neurological abnormality. OMIM Database Entries- Arthrogryposis List (about this entry) |
||||||
|
Fusion of fingers or toes. This may be single or multiple and may affect (i) Skin only (ii) Skin and soft tissues (iii) Skin, soft tissues and bone See Knock out Mouse Reference The condition is unimportant in toes but disabling in fingers and requires operative separation. This is frequently inherited as an autosomal dominant. Photo OMIM Database Entry- Syndactyly I |
||||||
|
This is involved with assymetric growth impairment of the vertebral bodies. There is lateral deviation of the spine with a 3-fold deformity:
Photos: Sco 1: Shows Scoliosis Sco 2: Rotational deformity producing rib hump when the child bends Sco 3: X-ray of spine The deformity is compensated by movement of the vertebral column above and below the affected region producing a primary and two secondary curves. This deformity progresses rapidly in adolescence and becomes fixed once bone growth is completed.
Questions Why does the deformity progress rapidly in early adolescence? |
||||||
|
CONGENITAL LIMB REDUCTION DEFECTS Thalilomide was the most celebrated limb reducing insult in humans which produced other deformities as well. There was probably a primary neuronal deprivation. There are two elements in the production of limb reduction defects. (a) Agents - Many substances have been found capable of producing limb reduction defects in experimental animals but few have been related to humans. Limb reduction defects may be apical (congenital amputation) or pre- or post-axial (absence of radius and lateral digits; ulnar and medial digits). Photo LRD 1 and 2 shows a limb reduction defect and the accompanying X-ray.
Questions What area is missing in the reduced limb? What will be the relative growth rates of the right and left humeri in this child? Other examples of limb reduction defects are shown by :- Photos: LRD 3:A reduction defect, largely preaxial hemimelia LRD 4: An apical defect LRD 5: A severe apical defect, the lobster claw foot. Question What problems would a patient with the lobster-claw foot defect encounter when walking? |
||||||
Self Assessment QuestionsSelf Assessment Questions
What are the major causes and consequences of congenital dislocation of the hip and scoliosis? |
||||||
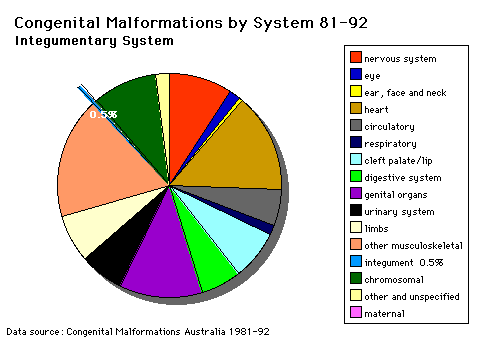
Integumentary System |
|
|||||
|
|
||||||
|
||||||
About Notes
|
|
|||||
|
m.hill@unsw.edu.au |
|
|||||